Pląsawica Huntingtona to ciężkie, genetycznie uwarunkowane, nieuleczalne schorzenie neurodegeneracyjne. Spowodowana jest nadmierną ilością powtórzeń sekwencji CAG (kodującej aminokwas glutaminę) w genie htt na chromosomie 4. Produktem takiego wadliwego genu jest białko huntingtyna (HTT) z dużo dłuższym niż u zdrowego człowieka łańcuchem glutaminowym, przy czym ilość powtórzonych glutamin bezpośrednio koreluje z prędkością wystąpienia objawów i ich ciężkością. Wśród objawów pląsawicy, obok charakterystycznych zaburzeń ruchowych, którym choroba zawdzięcza swoją nazwę, warto wymienić także zaburzenia kognitywne z postępującą demencją i zmiany osobowościowe.
Jakkolwiek wiadomo, że wadliwa huntingtyna uderza głównie w neurony (w chorobie obserwuje się utratę komórek nerwowych, zwłaszcza w obszarze mózgowia zwanym prążkowiem), mechanizm jej działania nie jest jasny. Liczne hipotezy dotyczące przyczyn jej neurotoksyczności od jakiegoś czasu szczególnie chętnie uwzględniały wpływ uszkodzonego białka na mitochondria. Mitochondria w komórkach pełnią funkcję niejako mikroelektrowni, zapewniając ciągły dopływ energii użytkowej do systemu i tym samym umożliwiając sprawne funkcjonowanie, tymczasem neurony to jedne z bardziej wymagających energetycznie komórek ludzkich - sprawna praca mitochondriów jest w nich szczególnie istotna. Wbrew temu, co mogłoby się wydawać komuś po krótkim kursie biologii w szkole (o ile w ogóle zapamiętał fakt istnienia takich struktur), mitochondria to organella niezwykle dynamiczne, potrafią wędrować po całej komórce, podlegają nieustającym cyklom fragmentacji i fuzji, tworząc coś w rodzaju komórkowej sieci energetycznej. Ich mobilność i plastyczność są szczególnie istotne w neuronach właśnie, gdzie, wytwarzane w samym centrum, w ciele komórki nerwowej, transportowane są później wzdłuż jej wypustek, by gromadzić się w pobliżu synaps, a ich gęstość, rozmiary i rozmieszczenie podlegają ciągłej, ścisłej kontroli. Zakłócenie tych procesów skutkuje zaburzeniami aktywności synaptyczej, utratą wypustek i w końcu śmiercią komórek. Poszczególne etapy rozregulowania dynamiki mitochondrialnej można obserwować nawet mikroskopowo.
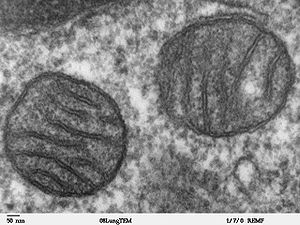
Mikrofotografia elektronowa dwóch mitochondriów
|
Wyniki badań opisujących wpływ huntingtyny na procesy fuzji i fragmentacji mitochondriów ukazały się w marcowym "Nature Medicine". Białkami niezbędnymi do regulacji łączenia się i rozpadu mitochondriów są enzymy z grupy GTP-az należące do rodziny tzw. dynamin, spośród których DRP1 jest czołowym związkiem odpowiedzialnym za proces fragmentacji. Właśnie z tym białkiem, jak odkrył zespół naukowców, defektywna huntingtyna potrafi się bezpośrednio wiązać, znacząco wzmagając jego aktywność, co zaburza delikatną wewnątrzkomórkową równowagę układu mitochondriów. Nadmierne pofragmentowanie mitochondriów przeszkadza w prawidłowym ich transporcie wenątneuronalnym i zmniejsza przeżywalność komórek.
Badacze pracowali na hodowlach komórek ludzkich (głównie fibroblastów uzyskanych od pacjentów z chorobą Huntingtona) i neuronów szczurzych (z sztucznie wywołaną ekspresją zmodyfikowanej huntingtyny) oraz na mysich mutantach produkujących różnej długości HTT. Już wstępne obserwacje morfologiczne "uszkodzonych" komórek nerwowych szczurów wykazały znaczące różnice w porównaniu z komórkami zdrowymi - mitochondria w próbie kontrolnej wykazywały budowę typową (podłużny kształt) i przeciętną długość około 3200nm, podczas gdy w populacji badanej były wyraźnie krótsze i zaokrąglone, o długości zależnej od ilości powtórzeń glutaminy w zmienionym białku, dochodzącej do 800nm przy 97 powtórzeniach (w grupie kontrolnej przyjęto 17 powtórzeń). W neuronach ze zmienionym HTT zaobserwowano też obniżenie ogólnej liczby tych organelli oraz zwiększony poziom autofagii, a wzmożenie fragmentacji mitochondriów korelowało bezpośrednio z poziomem obserwowanej śmierci komórek nerwowych. Podobne zmiany w dynamice mitochondriów obserwowano też w hodowanych komórkach ludzkich. Fragmentacja organelli powodowała także wyraźne obniżenie zarówno stopnia, jak i tempa ich transportu wewnątrzkomórkowego, doprowadzając w przypadkach o ekspresjii huntingtyny z dłuższymi łańcuchami glutaminy do nieomalże jego wstrzymania (znów stopień zahamowania korelował z długością łańcucha aminokwasów). Obserwacje potwierdzone zostały testami na żywych mysich mutantach, przy czym zmiany były obserwowalne nawet jeszcze przed wystąpieniem objawów klinicznych.
Badania immunologiczne i enzymatyczne tkanki nerwowej chorych gryzoni oraz obserwacje odpowiednio wybarwionych komórek potwierdziły fakt bezpośredniego wiązania się huntingtyny z wspomnianym białkiem DRP1 (przy czym huntingtyna zmutowana charakteryzowała się znacznie zwiększonym do DRP powinowactwem) powodujące zwiększenie jego aktywności, przy czym warto podkreślić, że jednocześnie HTT nie wykazywała żadnego powinowactwa do białka mitofuzyny o przeciwnym działaniu (promującym fuzję mitochondriów). Jednocześnie badania prowadzone na liniach komórkowych wykazały, że podwyższenie produkcji mitofuzyny lub obniżenie ekspresji DRP1 w znacznym stopniu niwelują wpływ uszkodzonej huntingtyny na komórki nerwowe. Zjawisko wiązania DRP1 przez huntingtynę potwierdzono także w komórkach krwi i bioptatach tkanki mózgowej chorych na pląsawicę.

Tydzień Mózgu
Powyższe badanie wskazuje nowy, tym razem bazujący na patofizjologii schorzenia, punkt uchwytu dla terapii pląsawicy Huntingtona. System mitochondrialnej fragmentacji i fuzji, a konkretniej układ białek DRP1/mitofuzyna mogą w przyszłości stać się celem kolejnej generacji leków tudzież terapii genowej, która na polu chorób neurodegeneracyjnych zaczyna właśnie wchodzić w etap praktycznego wykorzystywania.
Źródło: Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related
protein-1 and increases its enzymatic activity. Song W, Chen J,
Petrilli A, Liot G, Klinglmayr E, Zhou Y, Poquiz P, Tjong J, Pouladi MA,
Hayden MR, Masliah E, Ellisman M, Rouiller I, Schwarzenbacher R, Bossy
B, Perkins G, Bossy-Wetzel E. Nat Med. 2011 Mar;17(3):377-82.
Omówienie
Przydatne
opracowanie na temat choroby Huntingtona (ogólnodostępne i po polsku)
[Paulina Łopatniuk]
Paulina Łopatniuk |


