
|
Science » » Priony. Czyżby nowożytny miecz Damoklesa? [4] Choroba Creutzfeldta-Jakoba (CJD) Opisana w 1921 r. przez niemieckich neurologów, byli to Hans G. Creutzfeldt i Alfons Jakob. Przeważnie występuje u osób w wieku około 60 lat, z częstością 1 przypadku/milion/rok. [ 42 ] Wyróżniono postaci: sCJD (samoistna, występuje sporadycznie), jCJD (przepasażowna/jatrogenna), fCJD (rodzinna), vCJD (wariant CJD). Istotną cechą tej choroby jest to, że nie pojawia się skierowana przeciwko patogenom reakcja immunologiczna, bowiem układ odpornościowy nie rozpoznaje czynnika odpowiedzialnego za chorobę. Wynika to z faktu, że patogenem jest białko, naturalnie występujące w organizmie ludzkim. 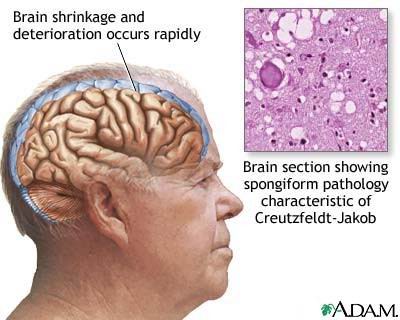 [ 43 ] [ 43 ]CJD W obrazie klinicznym w blisko 70 procentach przypadków występuje triada objawów: postępujące otępienie psychiczne, mioklonie [ 44 ], typowy zapis EEG. U około 30 procent chorych rejestruje się objawy prodromalne: osłabienie, zaburzenia snu (bezsenność, rzadziej nadmierna senność) i jedzenia. W co piątym przypadku choroba zaczyna się nagle. U 2/3 pacjentów występują zaburzenia zachowania i deterioracja intelektualna (pogorszenie się, psucie się, utrata wartości, spadek jakości). Także u 2/3 obecne są zaburzenia neurologiczne (chodu, podwójne widzenie bądź utrata wzroku, hemianopsja (połowiczna ślepota, ograniczenie pola widzenia do jednej połowy). Inne objawy: zaburzenia wyższych czynności nerwowych, palinopsje [ 45 ], mimowolne ruchy choreoataktyczne, objawy czuciowe potem zmiany neurologiczne (sztywność mięśniowa, drżenia, porażenie nerwów czaszkowych), po roku (u 90 procent przypadków), dwóch (niekiedy upływa dłuższy okres) następuje śpiączka i śmierć. [ 46 ] U 10-15 procent choroba ma charakter dziedziczny, jest wynikiem mutacji genu kodującego białko pionu. {P:47|Encyklopedia Multimedialna PWN' 2000 podaje jeszcze, że CJD wywoływana jest przez tzw. wirusy powolne, charakteryzuje ją bardzo długi okres inkubacji, do 30(40) lat (na pewno więcej niż 10 lat).} Patogenne priony obecne są w kępkach Periera jelita i wyrostka robaczkowego [ 48 ], migdałkach [ 49 ], układzie chłonnym/limfatycznym wraz z węzłami, śledzionie, płucach i sercu. Źródła nieumyślnych zakażeń: transplantacja rogówki (na świecie do 2000 r. odnotowano trzy przypadki), implantacja opony twardej i elektrod do mózgu (około stu przypadków), podskórne podanie hormonu wzrostu [ 50 ], badania endoskopowe, przeszczep wątroby lub błony bębenkowej, nieprawidłowo odkażone narzędzia chirurgiczne, uszkodzenia powłok ciała (rzeźnicy i in.). Badania eksperymentalne wskazują na możliwość zarażenia poprzez transfuzję zakażonej krwi. Nie jest znana dawka zakażająca [ 51 ] i wielkość populacji zakażonej. Ostatnimi czasy pojawiła się kontrowersyjna kwestia nowego wariantu, tzw. vCJD (przedstarcze otępienie umysłowe); został zidentyfikowany w roku 1996. Jest wynikiem przepasażowania encefalopatii gąbczastej bydła (BSE) na człowieka. Przy tym wariancie średni wiek zachorowania to 26 lat (przedział 19-41 lat); vCJD rozwija się do roku (7,5-23 mies.). Obraz kliniczny: zaburzenia zachowania (lęk, agresja) jako objawy prodromalne, ciągłe dyzestezje i ból w stopach [ 52 ], ataksja (wcześnie), otępienie (późno). Brak typowego EEG. Prawdopodobnie upośledzenie mowy przy kuru i CJD nie wynika z bezpośrednich uszkodzeń wyższych ośrodków mowy w mózgu, lecz jest skutkiem zaburzenia funkcji móżdżku, zwłaszcza synchronizacji czynności aparatu oddechowego, krtani, podniebienia, języka i ust, niezbędnej do prawidłowej fonacji. Upośledzeniu ulegają też działania móżdżku związane z zachowaniem równowagi. Syndrom Gertsmanna-Strausslera-Scheinkera (GSS) Objawy: ataksja oraz inne symptomy wskazujące na uszkodzenie móżdżku. Śmiertelna dziedziczna/rodzinna bezsenność (FFI) Objawy: po okresowych trudnościach w zasypianiu następuje otępienie. Początek w wieku 37-61 lat, trwa średnio 13 miesięcy (7-25 miesięcy). Została opisana w przypadku jednej 6-pokoleniowej rodziny. FFI występuje w równych proporcjach niezależnie od płci. Obraz kliniczny: szybko postępująca bezsenność, zaburzenia autonomiczne (nadmierna potliwość, podwyższona temperatura, nadciśnienie) i endokrynologiczne (zniesienie dobowego rytmu wydzielania somatotropiny [ 53 ] oraz stale podwyższony poziom kortyzolu przy obniżonym poziomie hormonu adrenokortykotropowego [ 54 ], ataksja, mioklonie, objawy piramidowe, demencja. Syndrom oraz bezsenność należą do chorób dziedzicznych, występują u osób w wieku średnim. Rodzinna postępująca glioza podkorowa Choroba charakteryzuje się nadmiernym rozrostem astrocytów — największe komórki glejowe, sieć ich wypustek stanowi zrąb dla układu nerwowego — w obrębie kory mózgowej z jednoczesną atrofią mózgu. Przyczyna choroby pozostaje nieznana. Klinicznie manifestuje się postępującym otępieniem bez istotnych zmian neurologicznych. Opisano kilka przypadków choroby o długoletnim przebiegu do 20 lat. Może przypominać inne szczególnie postępujące porażenie nadjądrowe. [ 55 ] Zespół Alpersa — in. choroba Alpersa (Alpersa-Huttenlochera) Rzadka, postępująca, uwarunkowana genetycznie choroba neurodegeneracyjna ośrodkowego układu nerwowego, o początku w okresie niemowlęcym lub wczesnym dzieciństwie. Może być spowodowana mutacjami w locus 15q25 w genie kodującym mitochondrialną polimerazę gamma. Dziedziczona w sposób autosomalny recesywny. Chorobę opisał jako pierwszy Alfons Maria Jakob (1884-1931), a publikacje na jej temat przedstawili trzej jego studenci: Souza, Freedomi Bernard Jacob Alpers (1900-1981). Kolejny opis zawdzięczamy duńskim neurologom — Erna Christensen i Knud Haraldsen Krabbe (1949 r.). Eponim medyczny (termin używany w medycynie) choroby Alpersa wprowadzono w 1963 roku. [ 56 ] Choroby zwierzęce:Scrapie (z ang. drapanie się) — in. kołowacizna, trzęsawka owiec. Choroba znana od XVIII wieku (opisana w Szkocji), miała początkowo charakter endemiczny. Objawy: utrata koordynacji ruchowej, pobudliwość, intensywne swędzenie prowadzące do wydrapywania sobie przez zwierzę wełny lub sierści. Występuje zakażenie boczne (owce zakażają się wzajemnie, nie wiadomo jak), ponadto przekazywanie pionowe owca ----à jagnię, przypuszczalnie w trakcie porodu. [ 57 ] W latach 30. XX w. okazało się, że scrapie jest zaraźliwa — kilkunastu tysiącom owiec islandzkich podano szczepionkę uzyskaną na bazie wyciągu z mózgów owiec szkockich. W ciągu kilku lat padło 1,5 tys. zaszczepionych sztuk (stąd też miejscowy weterynarz Sigrudsson sformułował hipotezę o istnieniu powolnych chorób wirusowych, rozwijających się dopiero w kilka lat po zakażeniu). [ 58 ] Zakaźna encefalopatia norek (TME) Przewlekła, wyniszczająca choroba jeleni {P:59|W Wyoming (USA) wśród jeleni choroba jest bardzo silnie zakaźna; 30 procent populacji stanowej jest zakażone. Nie wiadomo jakie są potencjalne skutki tego zjawiska dla ludzi.} i antylop.
Footnotes: [ 42 ] W
Polsce na 2000 r. nie opisano żadnych wariantów CJD natomiast stwierdza się
ok.12 przypadków choroby rocznie (teoretycznie powinno ich być około 40). [ 43 ] źródło:
http://www.pawelmazur.org/ [ 44 ] Mioklonie
— in.
zrywania mięśniowe powstałe przez zmiany o różnym umiejscowieniu (rdzeń kręgowy,
pień mózgu, kora mózgu i ośrodki podkorowe). Istotą zaburzeń są krótkotrwałe
kurcze (szarpnięcia) pojedynczego mięśnia lub całych grup mięśniowych. [ 45 ] Palinopsja
(perseweracja wzrokowa) — zaburzenie neurologiczne polegające na przetrwałym
widzeniu lub wielokrotnym pojawianiu się obrazu, mimo ustania działania
wywołującego go bodźca. [ 46 ] 5
procent przeżywa do 2 lat, a kolejne 5 procent stanowią przypadki o przebiegu
powolnym. [ 48 ] W Wlk. Brytanii przebadano wyrostki
usunięte z powodu zapalenia w wybranej osobogrupie celem stwierdzenia, w którymś
patologicznego prionu, wówczas możliwa byłaby ocena zasięgu zakażonej grupy.
Czynnika nie wykryto w żadnej próbce, jednak wyliczenia statystyczne wskazują na
teoretyczne prawdopodobieństwo występowania w tym państwie od kilkuset do 140
tys. przypadków CJD! [ 49 ] W
Wlk. Brytanii usuwa się rocznie około 80 tys. migdałków, obecnie (począwszy od
2000 r.) przeprowadzane są analogiczne badania jak przy wyrostkach, osobogrupa
jest za to znacznie liczniejsza, bardziej przez to reprezentatywna dla populacji
krajowej. [ 50 ] Najwięcej zakażeń w Japonii i we
Francji, gdzie produkowano hormon wzrostu z przysadek mózgowych mimo, że wiadomo
już było o przypadkach przeniesienia CJD tą drogą. Według danych z maja
2001 r. znamy ponad 40 przypadków tego rodzaju. W jednym z wariantów choroby
zakaziły się dzieci, najmłodsza zmarła miała 13 lat! Pamiętając o czasie
inkubacji warto zapytać kiedy i czym się zakaziła? [ 51 ] Teoretycznie
prion z zainfekowanego pokarmu ma w organizmie człowieka do pokonania kilka
przeszkód, m.in. musiałby przedostać się z jelit do ukł. limfatycznego i śledziony, następnie do mózgu. [ 52 ] Dyzestezja — zaburzenie normalnego czucia (wrażliwości): 1. osłabienie wrażliwości, 2.
bolesna albo niewłaściwa wrażliwość, 3. chorobliwa wrażliwość (zwłaszcza u melancholików) na wpływy zewnętrzne. [ 53 ] ozn. GH; hormon wzrostu wydzielany
przez przysadkę mózgową [ 54 ] Kortyzol
(in. hydrokortyzon), to naturalny hormon steroidowy wytwarzany przez korę
nadnerczy, główny przedstawiciel
glikokortykosteroidów. Wywiera szeroki wpływ na metabolizm, w kulturze
popularnej bywa określany nazwą hormon stresowy na równi z adrenaliną.
Drugi związek to inaczej kortykotropina (skrót: ACTH) — hormon przysadki
mózgowej, pobudzający korę nadnerczy do wydzielania kortyzolu i wielu słabo
działających androgenów. Dla obu tych hormonów brak wahań rytmu dobowego
wydzielania. [ 57 ] Były
przypadki, gdzie chore sztuki zabijano i kremowano, pastwiska wypalano,
odczekiwano kilka lat po czym wprowadzano nowe, zdrowe owce. Choroba pojawiała
się ponownie. Gdzie przetrwał wektor (typowano m.in. kleszcze)? Jeden z naukowców wsadził mózg ze scarpie do dziurkowanej puszki po czym zakopał
ją na dwa lata w ogródku. Po tym okresie z treści pozostałej w puszce zrobił
ekstrakt i zakaził myszy scarpie. Jak długo cząstki te
zachowują właściwości infekcyjne? [ 58 ] O
istnieniu tajemniczego wirusa światu doniosła para francuskich badaczy J. Cuille i P.L. Chellew 1938 r. « (Published: 24-02-2011 Last change: 25-02-2011)
page 951 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||



 Nauczyciel, publikował w piśmie "Gameranking", współpracuje z miesięcznikiem "21. Wiek" (członek zespołu redakcyjnego). Autor książki "Cywilizacja traw". Pióro do wynajęcia.
Nauczyciel, publikował w piśmie "Gameranking", współpracuje z miesięcznikiem "21. Wiek" (członek zespołu redakcyjnego). Autor książki "Cywilizacja traw". Pióro do wynajęcia.
| [ Cooperation ] [ Advertise ] [ Map of the site ] [ F.A.Q. ] [ Store ] [ Sign up ] [ Contact ] The Rationalist © Copyright 2000-2018 (English section of Polish Racjonalista.pl) | ||